QM/MM Steered Molecular Dynamics (SMD)
Defining the Collective Variable (CV) for Hydride Transfer
Steered molecular dynamics (SMD) simulations are employed to explore potential structural variations along a pre-defined reaction coordinate or collective variable (CV). This technique is particularly useful for speeding up processes that might otherwise take a considerable amount of time to happen naturally. Subsequently, these generated configurations can serve two main purposes: they can shed light on the structural alterations relevant to the underlying reaction, and they can be employed to calculate free energy changes along the given CV.
How to choose a starting value for CV in SMD simulations?
SMD simulations will steer your chosen CV from the defined initial position to the final position. In our case, we know that the hyride will be transferred to N1 atom of the substrate from the N5 atom of the flavin cofactor. So, we know that steering of hydride should stop once it reached to its destination N1 atom of the substrate. However, the difficult question is, what should be the starting distance of hydride ion from the N1 atom to start steering? In our last 5 ps QM/MM production runs, we found that hydride atom has variable distances from the N1 atom, and it would be difficult to guess the distance minima. There could be multiple approaches to handle this particular question, one should take the average of the distances b/w hydride and N1 atom obtained from the QM/MM simulations, or it could be a plausible distance based on the existing studies where the distance between the hydride donor and acceptor is well defined. We had no experience of comparing these approaches, however, we believe that one should consider this question genuinely, as this could save significant amount of computation time for sampling the CV. We have chosen a different approach here, we ran a QM/MM scan along the N1-hydride distance using Qsite package (Gaussian can also be used). From the QM/MM scan, we found that the N1-hydride distance has an initial minimum around 2.0 Angstrom, hence we have chosen 2.0 Angstrom as our starting distance to steer hydride atom from N5 atom to N1 atom.
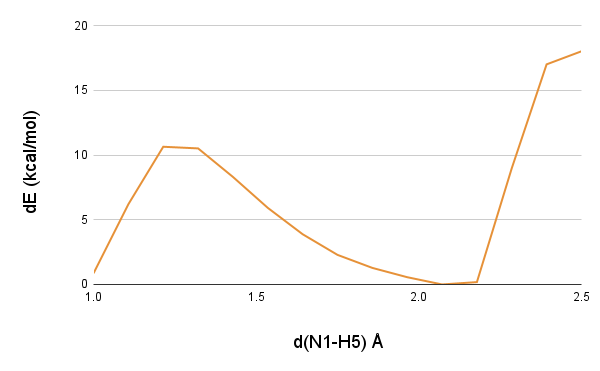
The 1D QM/MM scan along the distance b/w hydride receiver N1 atom and the hydride atom (H5)
In the context of our work, we are examining a sequential reaction involving an initial hydride transfer followed by a proton transfer. Guided by our QM/MM scan, we’ve determined that the hydride transfer occurs first, necessitating that we focus on steering the system along the CV corresponding to hydride transfer before turning our attention to the proton transfer CV. Details concerning our chosen CVs are provided in figure S27 of Sahrawat et al. (2024). Here is the CV file for the hyride transfer CV to be used for running QM/MM SMD simulations tutorial/simulations/mdin/cv-hy-1.in
cv_file
&colvar
cv_type = 'LCOD',
cv_ni = 4, cv_i = 11078,11007,10996,11007,
cv_nr = 2, cv_r = 1.0,-1.0,
npath = 2, path = 1.0,-1.0, path_mode = 'LINES',
nharm = 1, harm = 1000.0
/
The first highlighted line defines the atom numbers of the three atoms involved in hydride transfer. 11078 is substrates N1 atom, 11007 is the hyride ion (H5), 10996 is the flavin’s N5 atom. The hyride ion is being transferred from the N5 atom of the flavin to N1 atom of the substrate. The second highlighted line consist of the weightage factor for the respective distance, whereas the third highlighted line describe the starting and the end values of the handle position, respectively. The last highlighted line contains the value of spring constant. As we found out from the QM/MM scan that the starting value for the N1-H5 distance should be 2.0 A, taking this into consideration, our CV has a starting value of 1.0 Angstrom and will goes upto -1.0 Angstrom. Overall, we are steering H5 towards N1 atoms and at the same time away from the N5 atom of the flavin. The total displacement of H5 atom would be 1 Angstrom. Each SMD simulation will be of 1 ps (timestep = 1 fs), this means the velocity of steering the H5 atom is 1 Angstrom/ps. This is an acceptable velocity considering the time scale of hydride/proton transfer in proteins, which is in ps. So, we are aiming not to steer the hydride ion unrealistically and not too fast as well!
Here is the amber mdin file for running QM/MM SMD simulation along the hydride transfer CV. tutorial/simulations/mdin/qmmm-smd-hy-1.in
298K constant temp QMMMMD
&cntrl
imin= 0, ! Run molecular dynamics.
nstlim=1000, ! Number of MD-steps to be performed.
dt=0.001, ! Time step (ps)
ntb=2, ! Periodic conditiond at constant pressure
cut=8.0, ! non-bond cut off
ntc=2, ntf=2, ! Constrain lengths of bonds having hydrogen atoms (SHAKE) except flavin hydride HN5
irest=0, ig=-1, ! Generate a random seed for velocity
tempi=298.0, temp0=298.0, ! Temperature
ntt=3, gamma_ln=3.0, ! Temperature scaling using Langevin dynamics with the collision frequency in gamma_ln (ps−1)
ntp=1, taup=2.0, ! Pressure scaling
ntpr=1, ntwx=1, ntwr=1, ! Output options
ifqnt=1, ! Switch on QM/MM coupled potential
infe=1, ! Switch on free energy modules
/
&qmmm
qmmask = ':723|@10985-11005,11009-11010,11014-11015,11018-11023', ! Substrate and LumiFlavin atoms selected in the QM region
qm_theory = 'EXTERN', ! Opt for external QM software
qmcharge = -1, ! Total charge on the atoms defined in the QM regions
qmmm_int = 1, ! For Electronic embedding
qm_ewald = 0, ! Switch off Ewald summation and PME for QM-MM interactions
printcharges = 1, ! Option to print the atomic charges of QM atoms in mdout file
writepdb = 1, ! Write a pdb file showing the atoms selected in the SQM region, a good choice to verify selected atoms
verbosity = 1, ! Level of information to be printed in mdout for selected QM atoms
qmshake = 0, ! Turn off shake on QM atoms
/
&tc ! Syntax for using TeraChem as external QM software
method = 'B3LYP', ! Choice of QM theory
basis = '6-31G*', ! Basis set
guesss = 'scr/c0', ! SCF guess to read/write
scrdir = 'scr', ! Scratch directory
keep_scr = 'yes', ! Don't delete the content of scratch directory
ngpus = 2, ! Number of GPUs
gpuids = 0,1, ! Specify the GPU ids
use_template = 1, ! Read the TeraChem template file "tc_job.tpl"
/
&smd
output_file = 'smd-hy-1.txt'
output_freq = 1
cv_file = 'cv-hy-1.in'
/
We Recommend!
Chosing multiple starting configuration for SMD simulations!.
Now, given the stochastic nature of molecular dynamics simulations, it is generally advised to run multiple SMD trajectories from different initial configurations to better sample the reaction coordinate. We have chosen multiple starting configurations from our last three QM/MM production runs, where the hydride CV has a value of 1.0 Angstrom. As mentioned before, the value of 1.0 Angstrom is based on the fact that the coordinate scan along the hydride transfer CV has shown minimum at this distance, hence we have chosen this as our starting point for steering. Using VMD we have saved 10 random configurations (with CV=1.0) from the QM/MM production runs, and randomly using them as an input, we have 95 independent QM/MM SMD simulations for hydride transfer, followed by same number of simulations for proton transfer CV.
We have employed a bash script that will run the desired number of SMD simulations, while randomly choosing a starting configuration for each SMD run. Here is the content of the automated script tutorial/simulations/4-amber-tc-smd-hy-1.sh
#!/bin/bash
dir="qm_log"
if [ ! -d "$dir" ]; then
mkdir -p "$dir"
echo "Directory for storing QM log '$dir' created."
else
echo "Directory '$dir' already exists."
fi
for i in {1..95}
do
# Chose a random reference frame
j=$(shuf -i 1-10 -n1)
# Prefix for the input and output files
ref=step7.0.prod.hy.cv.2.0.${j}
step=step8.smd.hy.${i}
# Sander production run
sander -O -i mdin/qmmm-smd-hy-1.in -p xenA_h_OHP.parm7 -c ${ref}.rst7 -o ${step}.mdout -r ${step}.rst7 -inf ${step}.mdinfo -ref ${ref}.rst7 -x ${step}.nc &
sleep 5s
# Capturing QM log files at each step
count=0
# Whenever TeraChem completes its job, move the old log file to the qm_log directory
while ! grep "Final Performance Info" ${step}.mdinfo > /dev/null; do
if [[ -e old.tc_job.dat ]]; then
mv old.tc_job.dat qm_log/${step}_tc_${count}.dat
mv scr/charge_vdd.xls scr/${step}_charge_vdd_${count}.xls
((count=count+1))
fi
done
# Renaming SMD work record file for each step separately
mv smd-hy-1.txt smd-hy-1-${i}.txt
done
The first highlighted line defines that for each SMD run, a randomly selected input will be used from the 10 different starting configuration. This script will save and rename the charge_vdd.xls and tc_job.dat files at each step for each of the QM system. Later on, these files can be used to analyse the SCF, QM Energy, HOMO-LUMO Gap or the VDD charges etc.
The second highlighted line will save the CV-vs-work output from each SMD run, those are later to be used to compute the free energy profile along the CV. Finally, free energy profile along the CV have been calculated by the fluctuation-dissipation (FD) estimator, details of which is available in the supplementary information of Sahrawat et al. (2024) under section heading QM/MM Steered Molecular Dynamics.
Apart from these 95 SMD simulations, we have run 5 SMD simulations those are coupled with NBO analysis along the CV. Details of which is available in the next section.