Protein Parametrisation with AMBER
Important
Know your ligands and enzyme! This is very important before start following this or any other tutorials. Use any structural visualisation program like VMD, Pymol, Chimera, Maestro etc, to open input structural coordinate files. This will familiarise you with your input and will help you to track down any structural modelling deformity/error in subsequent processes. Especially, during force field (ff) parametrisation, where some programs guess bond order using supplied 3D coordinates and will assign ff parameter based on their guess. Be careful at this point! Sometimes a double bond might be treated as single or vice-versa.
Preparing Your Enzyme Structure for AMBER
Splitting the PDB in protein, ligand and water molecules
First, we will split the PDB into three groups: protein, ligands, and water molecules, as each requires separate treatment. OHP is the name of our oxime substrate; FMH is the flavin cofactor. Pay attention to the PDB Residue Name column (positions 18–20) for the name assigned to each molecule in the file.
We have modified the residue names of the flavin cofactor and the oxime substrate. If you want to do so, simply open the PDB file in a text editor and replace the existing name with a name of your choice (three characters or fewer). Be careful not to shift the columns, as this will corrupt the PDB field positions. Below are the bash commands to split the input PDB into protein, cofactor, substrate, and water files.
grep -v -e "FMH" -e "OHP" -e "CONECT" -e "HOH" 8AU8.pdb > protein.pdb
grep "FMH" 8AU8.pdb > FMH.pdb
grep "OHP" 8AU8.pdb > OHP.pdb
grep "HOH" 8AU8.pdb > waters.pdb
Assessing the protonation states of the enzyme with careful attention for active site residues
There are several methods to assess the protonation state of the residues in a protein. Not limited to following:
|
|
We Recommend!
H++ Server
The new release (version 4.0) of H++, uses AmberTools 20 to preprocess a PDB file and it is able to generate AMBER compatible PDB structure.
Warning
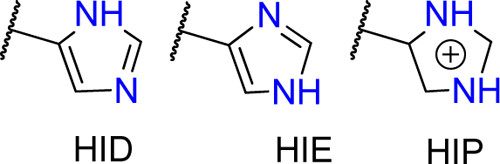
Histidines are tricky residues, they have three possible protonation states: HID, HIE and HIP. HIP is the protonated residue. HID and HIE correspond to the two natural tautomers of the neutral histidine, where the proton can be found in delta or epsilon positions. In solution, the most common conformer is HIE but always visualise your structure before assuming any histidine protonation state.
Here is an example showing the typical active site of Old Yellow Enzymes (OYE). The enzyme displayed here is XenA (an ene-reductase belongs to Old Yellow Enzyme 3) and after protonation, we found that protonation state of H178 predicted as HID, which is not correct, considering the presence of substrate, instead the NE atom of the H178 should have been protonated to form the hydrogen bond with the carbonyl oxygen of the substrate. Similarly, the ND atom of the H181 should be protonated instead of NE. Here, is the active site with correct protonation of H178 and H181.

Last but not least, we need to clean the PDB file before passing it to AmberTools (removing residues with double ocupation, checking for disulfide bonds, …). So we are going to use the first AmberTools tool: pdb4amber which prepares your PDB file.
pdb4amber -i protein.pdb -o protein4amber.pdb
Setting Histidine Protonation State with AMBER LEaP
We are using Ambertools to choose the desired protonation form of H178. You can select the appropriate form of HIS by renaming HIS to HIE (proton on NE2), HID (proton on ND1), or HIP (both protons). This way AMBER will add the required missing atoms for the specified protonation state. Be careful: if you want to mutate a residue (e.g., Lysine to Glutamic Acid), first delete the sidechain atoms of the Lysine from the PDB file before running the command — AMBER will then add the coordinates of the missing atoms for Glutamic Acid. In short, the trick is to retain only backbone atoms before any substitution.
module load amber/20
tleap -f leaprc.protein.ff14SB
prot = loadpdb protein4amber.pdb
set {prot.178 prot.181} name "HID"
savepdb prot wt_protein.pdb
quit
mol new protein.pdb
set prot [atomselect top "resid 177"]
$prot set resname HIS
set prot [atomselect top all]
$prot writepdb wt_protein.pdb
quit