System Equilibration and Minimisation
Removing bad atomic contacts and minimizing vacuum!
We have just built a “matured” system — all required molecules placed inside a simulation box. This step-wise assembly can introduce overlapping atomic positions, overly close contacts, or unrealistic separations between atoms, which in turn can cause artificial repulsion or prevent expected interactions.
Energy minimisation is therefore a crucial step before proceeding further. Using the potential energy terms from the force field, AMBER optimises atomic positions to resolve any clashes or vacuum artefacts in the initial configuration.
Briefly, the system will undergo sequentially through steps:
- Classical Energy Minimization
Will try to optimize the position of atoms using the supplied molecular mechanics parameters e.g. bond-length, angle, van-der-waal etc
Minimisation of system
&cntrl
imin=1, ! Perform an energy minimization.
maxcyc=4000, ! The maximum number of cycles of minimization.
ncyc=2000, ! The method will be switched from steepest descent to conjugate gradient after NCYC cycles.
ntr=1, ! Enabling restraints
restraint_wt = 10, ! 10 kcal/mol/A**2 restraint force constant
restraintmask = '!@H=&!:WAT,Na+' ! Restraints on the solute heavy atom
/
We are aiming to preserve the heavy atom positions, for this we are using a small restraint (10 kcal/mol/A^2) on the protein’s heavy atom. You can comment out the highlighted lines (just add ! at the beginning) if you want to minimise all atoms.
- Thermalisation
Kinetic energy or say the dynamics of the atoms increased step-wise
Heating ramp from 0K to 298K
&cntrl
imin=0, ! Run molecular dynamics.
ntx=1, ! Initial file contains coordinates, but no velocities.
irest=0, ! Do not restart the simulation, (only read coordinates from the coordinates file)
nstlim=15000, ! Number of MD-steps to be performed.
dt=0.002, ! Time step (ps)
ntf=2, ntc=2, ! Constrain lengths of bonds having hydrogen atoms (SHAKE)
tempi=0.0, temp0=298.0, ! Initial and final temperature
ntpr=500, ntwx=500, ! Output options
cut=8.0, ! non-bond cut off
ntb=1, ! Periodic conditiond at constant volume
ntp=0, ! No pressure scaling
ntt=3, gamma_ln=2.0, ! Temperature scaling using Langevin dynamics with the collision frequency in gamma_ln (ps−1)
ig=-1, ! seed for the pseudo-random number generator will be based on the current date and time.
ntr=1, ! Turn on positional restraints
restraint_wt = 10, ! 10 kcal/mol/A**2 restraint force constant
restraintmask = '!@H=&!:WAT,Na+' ! Restraints on the backbone heavy atom
nmropt=1, ! NMR options to give the temperature ramp.
/
&wt type='TEMP0', istep1=0, istep2=10000, value1=0.0, value2=298.0 /
&wt type='TEMP0', istep1=10001, istep2=15000, value1=298.0, value2=298.0 /
&wt type='END' /
This is a 30 ps NVT equilibration where the temperature is linearly increased from 0 to 298 K over the first 20 ps and for the rest of 10 ps, temperature will remain constant at 298K. Let’s see if this is true or not!
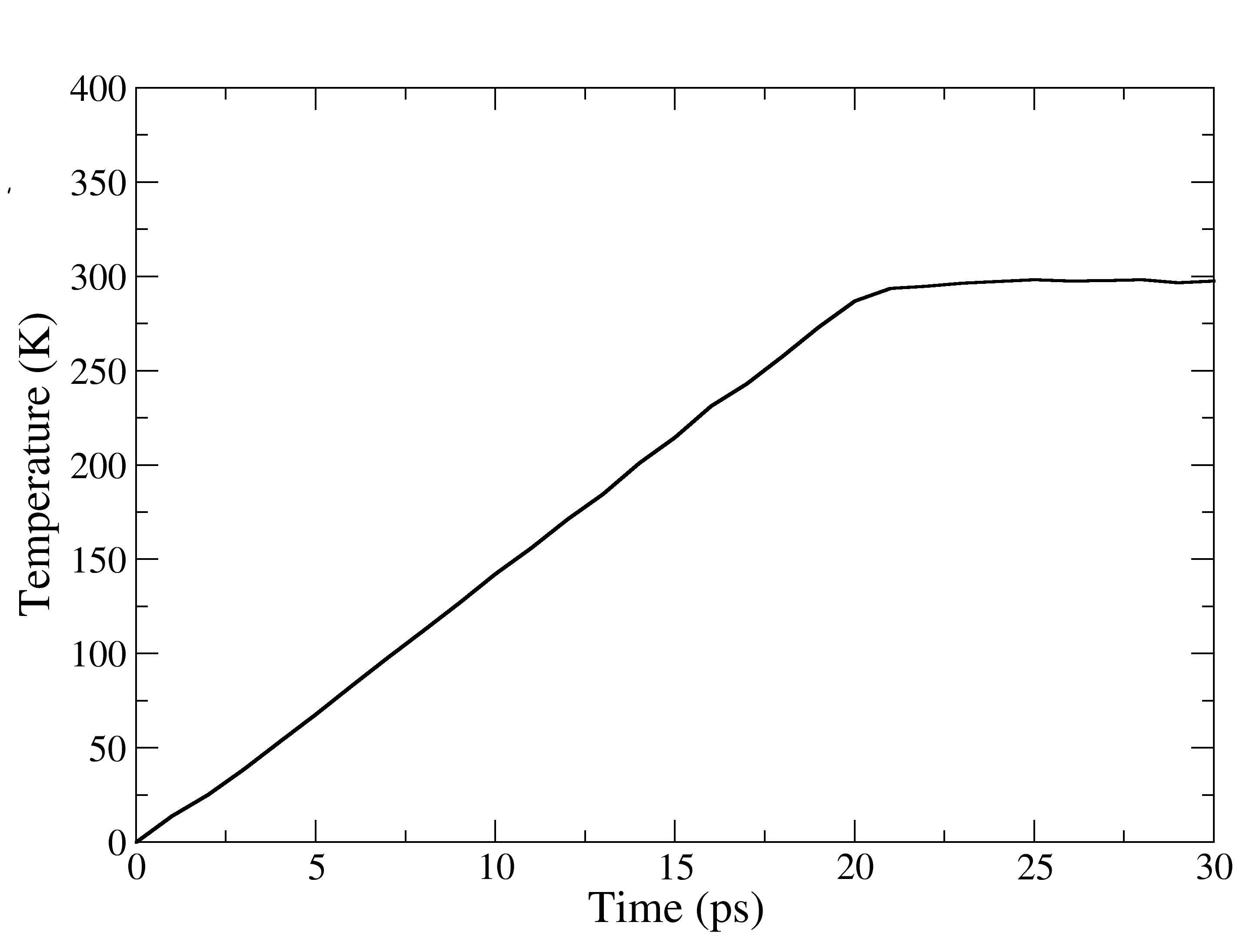
Figure showing the changes in the temperature vs Time (ps), during thermalisation. Upto 20 ps its an linear increase thereafter remained at 298K.
- Equilibration Run
Allowing the system to breathe for 100 ps, more precisely no restraint!
Density equilibration
&cntrl
imin= 0, ! Run molecular dynamics.
nscm= 1, ! Remove translational motion after specified steps
nstlim=50000, ! Number of MD-steps to be performed.
dt=0.002, ! Time step (ps)
irest=1, ! Restart the simulation and read coordinates
ntx=5, ! Initial file contains coordinates and veloci
ntpr=500, ntwx=500, ntwr=500, ! Output options
cut=8.0, ! non-bond cut off
temp0=298, ! Temperature
ntt=3, gamma_ln=3.0, ! Temperature scaling using Langevin dynamics
ntb=2, ! Periodic conditiond at constant pressure
ntc=2, ntf=2, ! Constrain lengths of bonds having hydrogen a
ntp=1, taup=2.0, ! Pressure scaling
iwrap=1, ioutfm=1, ! Output trajectory options
ntr=1, ! Enabling restraints
restraint_wt = 10, ! 10 kcal/mol/A**2 restraint force constant
restraintmask = '!@H=&!:WAT' ! Restraints on the solute heavy atom
/
This is a usual NPT run for 100 ps. Just to allow the solvent and hydrogen atoms to explore their neighbourhood.
- SQM-MM Energy Minimization
A part of the system treated with Semi-Empirical Method, rest of the system still under the classical ff
Energy minimisation using SQMMM
&cntrl
imin=1, ! Perform an energy minimization.
maxcyc=1000, ! The maximum number of cycles of minimization.
ncyc=500, ! The method will be switched from steepest descent to conjugate gradient after NCYC cycles.
ntb=1, ! Periodic conditiond at constant volume
cut=8.0, ! 8 angstrom classical non-bond cut off
ntpr=100, ntwx=100, ! Output options
ntc=2, ntf=2, ! Constrain lengths of bonds having hydrogen atoms (SHAKE)
ifqnt=1 ! Enable Quantum Module
/
&qmmm
qmmask = ':723|@10985-11005,11009-11010,11014-11015,11018-11023,416-429,2630-2640,2677-2687,2701-2715', ! Include the full side-chain of Y27,H178,H181,Y183, Lumiflavin and oxime',
qm_theory = 'PM6-DH+', ! Name of SQM method to use, please look in amber manual for available options
qmcharge = -1, ! Total charge on the atoms defined in the SQM regions
qmmm_int = 1, ! For Electronic embedding
qm_ewald = 0, ! Switch off Ewald summation and PME for SQM-MM interactions
writepdb = 1, ! Write a pdb file showing the atoms selected in the SQM region, a good choice to verify selected atoms
/
The flag “qmmask” up here, is defining the region to be treated using SQM. It is highly recommended to turn on the “writepdb” flag. The “writepdb” flag would write a pdb file named “qmmm_region.pdb” for the defined SQM/QM region, which should be opened in a molecule viewer and should be checked for the correctness.
Important
Check your paths!!
We are going to run a pipline, where Amber, a QM package (TeraChem or Gaussian ) and later NBO programs are interconnected, you should have these programs installed and their correct paths should be in your .bashrc or .bash_profile. Or you can add their paths in each of your automated bash scripts utilising them. Otherwise, this pipepline would not work and will display errors like sander not found!! or not binaries available for Gaussian or TeraChem etc. We recommend adding these line in your .bashrc:
# Source Amber, TeraChem and NBO source $your amber installation directory$/amber.sh
source $your terachem installation directory$/SetTCVars.sh
export NBOEXE=”$your terachem/nbo installation directory$/bin/nbo6.i4.exe”
- QM-MM Energy Minimization
A part of interest uses QM and rest is still under classical ff
We have used an external QM package for the QM calculations, although AmberTools22/23 has an intergated DFT package named QUICK
We have used TeraChem, a QM package fully operational on GPU. We have used demo version of it, which supports upto two GPUs and a maximum 15 mins of runtime for a given calculation. The given constraint on demo version of TeraChem was sufficient to run our QMMM MD simulations
Amber supports a wide range of QM package for QMMM simulations like Gaussian, Gamess, Orca etc. For more info please visit the Amber manual
Energy minimisation using QMMM
&cntrl
imin=1, ! Perform an energy minimization.
maxcyc=500, ! The maximum number of cycles of minimization.
ncyc=250, ! The method will be switched from steepest descent to conjugate gradient after NCYC cycles.
ntb=1, ! Periodic conditiond at constant volume
ntpr=50, ntwx=50, ! Output options
cut=8.0, ! 8 angstrom classical non-bond cut off
ntc=2, ntf=2, ! Constrain lengths of bonds having hydrogen atoms (SHAKE)
ifqnt=1 ! Enable QM-MM
/
&qmmm
qmmask = ':723|@10985-11005,11009-11010,11014-11015,11018-11023,416-429,2630-2640,2677-2687,2701-2715', ! Include the full side-chain of Y27,H178,H181,Y183,Lumiflavin and oxime',
qm_theory = 'EXTERN' ! Opt for external QM software
qmcharge = -1, ! Total charge on the atoms defined in the QM regions
qmmm_int = 1, ! For Electronic embedding
qm_ewald = 0, ! Switch off Ewald summation and PME for QM-MM interactions
printcharges = 1, ! Option to print the atomic charges of QM atoms in mdout file
writepdb = 1, ! Write a pdb file showing the atoms selected in the SQM region, a good choice to verify selected atoms
verbosity = 1, ! Level of information to be printed in mdout for selected QM atoms
qmshake = 0, ! Turn off shake on QM selected QM atoms
/
&tc ! Syntax for using TeraChem as external QM software
method = 'B3LYP', ! Choice of QM theory
basis = '6-31G*', ! Basis set
ngpus = 2, ! Choice for number of GPUs to use for TeraChem
gpuids = 0,1, ! Specify the GPU id's
use_template = 0, ! No template specified for TeraChem input
/
Note
We have used a script to automtise the above steps. If you are following these steps for the first time, do not run this script blindly. Be aware that it depends on the system to system, how much and which equilibration you need. Especially for a completely user-build system, unlike a crystal structure you need extra equiliration time.
Here is the content of the full automated script tutorial/pre-processing/1-amber-pre-run.sh
#!/bin/csh
# Written By Amit Singh. This script is for the automation of amber simulations
# Please change the variable below according to your system
set system = xenA_h_OHP
set amber = pmemd.cuda
set init = xenA_h_OHP
# These are just naming convention for different steps we are going to use
set mdin_prefix = mdin
set mini_prefix = step1.0_mm_mini
set heat_prefix = step2.0_thermalisation
set equi_prefix = step3.0_equilibration
set sqm_prefix = step4.0_sqm_min
set qmmm_prefix = step5.0_qmmm_min
# Step 1 --> Classical Minimization
# If there is a problem during minimization using pmemd.cuda, please try to use pmemd only for
# the minimization step.
if ( ! -e ${mini_prefix}.rst7 ) then
${amber} -O -i ${mdin_prefix}/mm_min.in -p ${init}.parm7 -c ${init}.rst7 -o ${mini_prefix}.mdout -r ${mini_prefix}.rst7 -inf ${mini_prefix}.mdinfo -ref ${init}.rst7
endif
# Step 2 --> Thermalisation
if ( ! -e ${heat_prefix}.rst7 ) then
${amber} -O -i ${mdin_prefix}/mm_heat.in -p ${init}.parm7 -c ${mini_prefix}.rst7 -o ${heat_prefix}.mdout -r ${heat_prefix}.rst7 -inf ${heat_prefix}.mdinfo -ref ${mini_prefix}.rst7 -x ${heat_prefix}.nc
endif
# Step 3 --> A Short 100ps Equilibration Run
if ( ! -e ${equi_prefix}.rst7 ) then
${amber} -O -i ${mdin_prefix}/mm_equil.in -p ${init}.parm7 -c ${heat_prefix}.rst7 -o ${equi_prefix}.mdout -r ${equi_prefix}.rst7 -inf ${equi_prefix}.mdinfo -ref ${heat_prefix}.rst7 -x ${equi_prefix}.nc
endif
# Step 4 --> Energy Minimization using Semi-Empirical Appraoch (SQM-MM)
if ( ! -e ${sqm_prefix}.rst7 ) then
sander -O -i ${mdin_prefix}/sqm_min.in -p ${init}.parm7 -c ${equi_prefix}.rst7 -o ${sqm_prefix}.mdout -r ${sqm_prefix}.rst7 -inf ${sqm_prefix}.mdinfo -ref $#{equi_prefix}.rst7 -x ${sqm_prefix}.nc
endif
# Step 5 --> Energy Minimization using Quantum Mechanical Method (QM-MM)
if ( ! -e ${qmmm_prefix}.rst7 ) then
sander -O -i ${mdin_prefix}/qmmm_min.in -p ${init}.parm7 -c ${sqm_prefix}.rst7 -o ${qmmm_prefix}.mdout -r ${qmmm_prefix}.rst7 -inf ${qmmm_prefix}.mdinfo -ref ${sqm_prefix}.rst7 -x ${qmmm_prefix}.nc
endif
Through the above script we have equilibrated the solvent positions around the enzyme, followed by minimisation steps using QM/MM. We will use the QM/MM minimised structure from step5 for all further investigation.